|
Bonds
|   |
|
Bonds
| |
Bonds between atoms are characterized by a conventional bond order which accounts for different bond lenghts.
Conventional bond order takes the following values:
Several molecular descriptors encode information on bond multiplicity by using the conventional bond order. They are thus affected by the conventional bond order assigned to molecules bonds.
When DRAGON reads a molecule input file, initial bond order values are assigned to molecule bonds according to the specific information contained in the input file. Single, double and triple bonds are specified in all the input files, excluding SMILES notations where single bonds are usually omitted. Aromatic bonds may be specified by specific symbols, e.g. "a" in HyperChem files, "Ar" in MDL and Sybyl files, ":" in SMILES notations. In SMILES notations, aromatic bonds are usually omitted, nonetheless DRAGON assigns bond order of 1.5 to bonds between two atoms entered by lower case atomic symbols. In Sybyl files, bonds represented by the symbols "am" and "du" are always assigned a bond order of 1.
At the stage of importing of molecule input files, the user can decide whether to keep the bond orders specified in the input file or to change them according to some rules implemented in DRAGON. In order to keep the original bond orders, the checkbox "User defined bond orders" in the "Select files" window must be checked. Note that this option is available only for DRAGON run in stand-alone mode. When DRAGON is running in background mode the bond orders are always recalculated except when the molecules are entered in the H-depleted form.
The DRAGON algorithm for conventional bond order rearrangement has been developed with the main goal of assigning value of 1.5 only to bonds belonging to effective aromatic structures (see rules for aromaticity detection). Examples are:
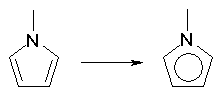
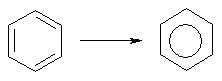
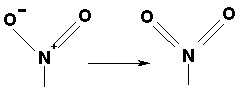
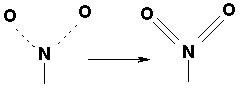
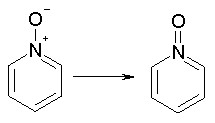
Delocalized bonds, such as in the nitro group, are represented as covalent bonds to both uncharged oxygens (N = O); they are always assigned a conventional bond order of 2. Examples are:
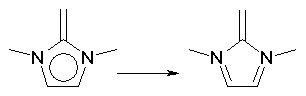
Moreover, if bond orders of 1.5 have been improperly assigned to non-aromatic structures, they are changed in such a way to obtain alternating single and double bonds. This correction is mainly concerned with HyperChem files and SMILES notations where the user has more liberty in explicitely defining aromatic bonds. Examples are:
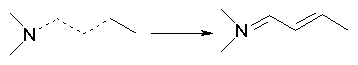
Some structures erroneously entered as aromatic by the user may be rejected if they are formally incorrect. This happens when alternating single and double bond assignments cannot be made. Example is:
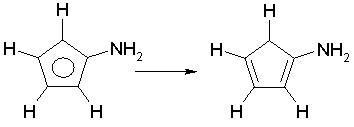
In the example above, there is no way to assign alternating single and double bonds without leaving one carbon atom with valence lower than 4.
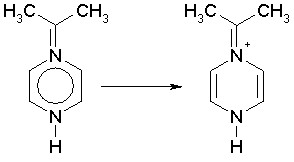
If a non-aromatic structure is entered as aromatic, it may also happen that different interpretations are possible. This is much more likely when the structure includes nitrogen atoms, which, having common valences of 3 and 5, but also valence of 4 in the case of a positively charged type (N+), can cause some ambiguities. An example is:
first solution
second solution
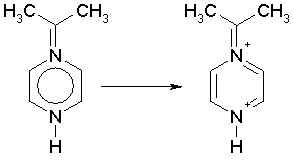
While in the first case one nitrogen is a positively charged type N+ and the other is a common nitrogen with valence of 3, in the second case one nitrogens is considered as N+ and the other as a nitrogen with valence of 5. The DRAGON algorithm would give the second solution because in the original structure both nitrogens have a valence greater than 3. DRAGON detects as N+ type any nitrogen with valence greater than 3 and lower than 5.
In reading SMILES notations DRAGON may initially assign several wrong bond orders of 1.5 if the user has entered atoms by lower case atomic symbols. Therefore, to avoid an erroneous molecule representation, the SMILES user is suggested to use lower case atomic symbols only for atoms effectively belonging to aromatic structures or to allow DRAGON to rearrange the conventional bond orders according to the internal algorithm for aromaticity detection.